Spinalna mišićna atrofija (SMA) je nasledna neurološka bolest koja uzrokuje progresivno slabljenje mišića.
U ovom tekstu koji je za vas napisala Milica Bogićević, molekularni biolog, saznajte:
Spinalna mišićna atrofija (SMA) je nasledna neurološka bolest koja uzrokuje progresivno slabljenje mišića. Bolest utiče na motorne neurone u kičmenoj moždini, što dovodi do smanjenja veličine i snage mišića. SMA se obično manifestuje u prvim mesecima života, ali se simptomi mogu pojaviti kasnije u životu.
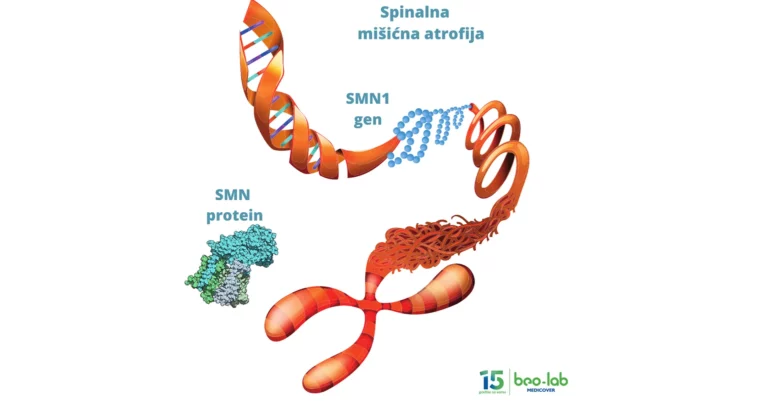
Genetički uzrok bolesti je mutacija u genu smn1 (Survival Motor Neuron 1), koji je odgovoran za proizvodnju proteina motorne jedinice kičmene moždine. Protein SMN (Survival Motor Neuron) je produkt smn1 gena i ključan je za preživljavanje motornih neurona i održavanje funkcionalnih veza između neurona i mišića.
Prvi simptomi Spinalne mišićne atrofije se mogu manifestovati u ranom detinjstvu, vreme pojave simptoma zavisi od tipa bolesti. Usled pomenutog oštećenja motornih neurona nastaje progresivna slabost mišića dalje praćena atrofijom mišića (smanjenjem mišićne mase) koje dovode do smanjene pokretljivosti i snage u zahvaćenim delovima tela. Bolest može zahvtatiti bilo koju grupu skeletnih mišića, te se često sreću gubitak mišićnih rekleksa, otežano disanje i gutanje usled oštećenja motornih neurona dijafragme i mišića grla.
Klasifikacija SMA se vrši na osnovu trenutka pojave prvih simptoma bolesti. Prema kliničkoj klasifikaciji postoje čeriti tipa SMA, koji će biti detaljnije opisani.Postoji peti (nulti (0)) tip SMA, koji se završava intrauterinom smrću ili par dana nakon rođenja zbog slabih refleksa, otežanog disanja i gutanja. Nutli tip se veoma retko javlja.
Tip I:
SMA tip I se javlja u prvim mesecima života sa simetričnom slabošću ekstremiteta, odsustvom refleksa tetiva, slabošću interkostalnih mišića i spazmom jezika. Prisutna je slabost mišića grudnog koša što uzrokuje otežano disanje i kašalj. Slabi mišići vrata onemogućavaju podizanje glave, samostalno sedenje, otežano je pomeranje ruku,prstiju i podizanje nogu. Ukoliko se ne leče, pacijenti sa SMA tip I ne postižu sposobnost sedenja. Očuvana je mimika lica, te se deca mogu smejati i mrštiti jer mišići lica nisu ozbiljno pogođeni.
Ishrana je veoma otežana i često se javlja neuspeh u napredovanju što je praćeno poremećajem gastrointestinalne peristaltike,dok otežano gutanje nosi veliki rizik da hrana ili tečnost završe u disajnim putevima. Pacijenti imaju očuvanu inteligenciju iako je mogućnost pokreta ograničena ili potpuno odsutna.
Tip II:
SMA tip II se javlja između šestog i osamnaestog meseca starosti, deca postižu sposobnost sedenja ali ne mogu samostalno da stoje ili hodaju. Prisutna je slabost i otežano pomeranje mišića ruku sa minimalnim pokretima prsitiju. Deca imaju poteškoće sa podizanjem nogu koje su slabije u odnosu na ruke. Kontrolisano mokrenje i pražnjenje creva nije zahvaćeno i vrši se bez poteškoća. Zbog slabosti mišića kičmenog stuba deca imaju skoliozu (bočno krivljenje kičme), takođe se vremenom zbog slabog kretanja javljaju kontrakture (ograničena pokretljivost zglobova).
U odnosu na tip I, reč je o lakšem obliku ali I dalje postoji problem sa disanjem i većina pacijenata zahtevapotporu disanja. Javljaju se poteškoće u ishrani i gutanju Mnogi pacijenti razvijaju kifoskoliozu, koja pogoršava osnovne respiratorne poteškoće i dovodi do potrebe za korisćenjem ortopedskih pomagala.
Tip III:
SMA tip III se javlja nakon osamnaestog meseca, u odnosu na tip II, predstavlja blaži oblik. Obolela deca postižu samostalno hodanje, ali sposobnost može da se izgubi tokom vremena. (najčešće se dešava u srednjem životnom dobu). Bolovi u mišićima, zglobovima, umor i padovi pri aktivnostima su česti što zahteva pomoć u svakodnevnim aktivnostima. Kao propratni simptomi mogu se javiti i disfunkcija gutanja i/ili poteškoće u ishrani. Tokom odrastanja i u pubertetu deca su sklona gubitku sposobnosti pokreta koje su imali do tada, kao što su samostalno hodanje i penjanje uz stepenice. Kao I kod tipa II, veoma česti simptomi su skolioza i kontrakture.
Tip IV:
Prvi simptomi se javljaju u kasnijem životnom dobu, kod odraslih osoba, sa varijabilnošći početka motoričkih poteškoća. Karakteristični simptomi su
Kada se jave prvi simptomi vremenom postepeno napreduju i slabost mišića se povećava, što može uticati na svakodnevne životne aktivnosti kao što su hodanje, kupanje i oblačenje. Problemi sa gutanjem i disanjem se vrlo retko javljaju Pojava simptoma ne utiče na dužinu žitotnog veka, ali znatno narušava kvalitet života.
Za razumevanje nastanka naslednih bolesti potrebno je najpre objasniti šta je genetički materijala. Naše telo je sačinjeno od miliona ćelija koje sadrže genetički materijal, poznat kao DNK (dezoksiribonukleinska kiselina). Dužina jednog molekula DNK isnosi dva metra, zbog toga se u ćelijama kompaktno organizuje u strukturi poznatoj kao hromozom. Svaka ćelija sadrži 46 hromozoma, podeljenih u 23 para. Od tih parova, 22 para su autozomni (telesni) hromozomi, dok jedan par određujuje pol, polni hromozomi.

Slika 1. Hromozom je gusto pakovan molekul DNK. Gen je segment molekula DNK koji nosi informaciju za sintezu funkcionalnog produkta.
Genetički materijal nasleđujemo od roditelja, svaki hromozom dolazi u dve kopije, po jedna od svakog roditelja. Gen je segment DNK molekula koji se sastoji od niza gradivnih elemenata zvanih nukleotidi. Gen sadrže uputstvo za izradu funkcionalnih produkata, među kojima su ključni proteini. Proteini su neophodni za normalno funkcionisanje ćelija, osiguravajući njihovu stabilnost i vitalnost. Gen nije kontinuiran segment sastoji se od delova koji kodiraju proteine (egzoni) i nekodirajućih delova (introni).
Ukoliko dođe do promene, tzv. mutacije na nekom od gena, sami proteini mogu biti ’’pogrešno napravljeni’’, što dalje utiče i na njihovu funkciju. Ovakve promene mogu biti nasledne – ukoliko se prenose na potomstvo sa roditelja, ali i stečene, pod uticajem različitih faktora unutrašnje i spoljašnje sredine (npr. radijacija).

Slika 2: Nasledni materijal dete nasledjuje od roditelja, čiji je udeo podjednak.
Gen smn1 (Survival Motor Neuron 1) se nalazi na petom hromozonu, u oblasti označenoj “5q12-q13”. Kao i drugi geni, nasleđuje se u dve kopije, po jedna od svakog roditelja. Gen smn1 je odgovoran za proizvodnju proteina SMN (Survival Motor Neuron), koji je ključan za održavanje motornih neurona u kičmenoj moždini. Protein SMN obezbeđuje dovoljnu dužinu aksona koji je u interakciji sa ćelijama skeletnog mišića. Pored smn1, u istoj oblasti se nalazi i identičan gen smn2 koji takođe kodira protein SMN, ali proizvodi manje funkcionalan protein u poređenju sa smn1.

Slika 3: Protein SMN obezbeđuje dovoljnu dužinu motoneurona koji je u interakciji sa ćelijama skeletnog mišića Interakcija neurona i mišića obezveđuje pokretljivost skeletnih mišića
SMA se nasleđuje autozomno recesivno. To znači da će se bolest ispoljiti samo ako osoba nasledi dve kopije mutiranog gena smn1po jednu od svakog roditelja, koji su nosioci mutacije. Osoba koje naslede samo jednu kopiju mutiranog gena obično neće pokazivati simptome, ali će biti nosioci te mutacije sa rizikom da je prenesu na potomstvo.

Slika 4: Verovatnoća za nasleđivanje mutiranog rega I pojavu bolesti u potomstvu
Postavljanje rane dijagnoze SMA može biti brzo i pouzdano, sprovedeno kroz genetičko testiranje. Ključna stvar je identifikacija smn1 kao glavnog uzročnika SMA, otkriće mutacija koje su odgovorne za nastanak bolesti. Poznato je preko dvadeset mutacija u genu smn1, koje mogu biti uzročnik bolesti.
Prema tome genetičko testiranje je najpouzdanija metoda koja se primenjuje ukoliko osoba ima simptome ili pozitivnu porodičnu anamnezu za SMA. Rana dijagnostika prilikom prvih simptoma je od presudnog značaja za početak lečenja, kao i za otkrivanje nosilaca mutacije ukoliko se planira potomstvo.
Simptomi mišićne slabosti su vrlo slični među različitim neuromišićnim oboljenjima, pa se na osnovu kliničke slike ne može sa sigurnošću znati da li je reč od SMA ili nekom drugom oboljenju. Eelektromiografija (EMG) koja potvrđuje smanjenu provodljivost neurona, biopsija mišića prikazuje brojnost mišićnih ćelija, magnetna rezonanca (MR) i kompjuterizovana tomografija (CT) dalju detaljnu sliku unutrašnjeg stanja mišićnog i nervnog sistema. Definitivna potvrda dijagnoze SMA se vrši putem genetičkog testiranja koje identifikuje mutirani gen smn1.
Prenatalna dijagnostika omogućava rano otkrivanje mutacije smn1 gena tokom trudnoće. Postoji nekoliko metoda prenatalne dijagnostike za identifikovanje prisutne mutacije, kao što je amniocenteza, horionska biopsija i neinvazivni prenatalni test (NIPT).
Amniocenteza je postupak u kojem se uzima uzorak amnionske tečnosti koja okružuje fetus u materici, između 15. i 20. nedelje trudnoće. Horionska biopsija je postupak u kojem se uzima uzorak horionskih resica iz materične posteljice. Postupak se obično obavlja između 10. i 12. nedelje trudnoće. Uzorci amnionske tečnosti i horionskih resica se analiziraju kako bi se utvrdilo prisustvo gena, broj kopija i moguće mutacije u genu. Neinvazivni prenatalni test (NIPT) je test prilikom kojeg se uzorkuje majčina krvi kako bi se analizirala fetalna DNK. NIPT se obično obavlja od 10. nedelje trudnoće.
Sekvenciranje kompletne genomske regije koja sadrži smn1 i smn2, otkriva 99% homologije između dve kopije, smn1 se razlikuje od smn2 za pet nukleotidnih razlika što ne utiče na kodiranje funkcionalnog proteina. Nastanak mutacije ili delecije u smn1 genu dovodi do gubitka proteina SMN i nastanka bolesti. Smn2 proizvodi transkripte sa preskočenim egzonom 7 koji je ključan za funkcionalan protein koji se kodira i na taj način smanjuje verovatnoću sinteze SMN proteina. Količina funkcionalnih proteina kodiranih sa smn2 gena utiče na težinu simptoma bolesti.

Slika 5: Gen smn1 je odgovoran za proizvodnju funkcionalnog proteina SMN, gen smn2 ima istu ulogu ali proizvodi mnogo manju količinu funkcionalnog proteina.
SMN (Survival Motor Neuron), protein je polipeptid koji se nalazi u svim tkivima, dok u spinalnim motornim neuronima dostiže visok nivo. Nivo SMN proteina značajno je smanjen u tkivima pacijenata sa SMA tip I, dok je umereno smanjen u tkivima pacijenata sa tipovima 2 i 3.
Egzoni 6 I 7 u okviru gena smn1,2 sadrže funkcionalno značajne domene koji su odgovorni za potpunu funkcionalnost SMN proteina. Preskakanje eksona ili mutacije dovode do smanjenja sinteze funkcionalnog proteina.
Na molekularnom nivou SMA tip I nosi homozigotne delecije smn1 (delecije u obe kopije gena), dok tip 2 i 3 pokazuju homozigotno odsustvo smn1, praćeno konverzijom gena smn1 u smn2. Što više kopija gena smn2 postoji, to je prisutnija veća količina potpunog SMN proteina i blaži je fenotip SMA. U 95% slučajeva odsustvo smn1 gena dovodi do razvoja bolesti dok homozigotno odsustvo smn2 nema kliničke posledice po nosioca. Gubitak smn 1 je ključan za nastanak SMA, dok je težina bolesti uglavnom povezana sa brojem kopija smn 2.
Najčešći mehanizam kojim dolazi do odsustva smn1 gena vezan je za region 5q12-q13, gde su locirani geni smn1 i smn2. Visoka učestalost de novo (prvi pu se javlja) mutacija nastaje prilikom nejednake rekombinacije, takođe su identifikovane duplikacije i inverzije veličine od 500 kb kod većine ispitivanih pacijenata u ovom regionu.

Slika6: Mutacija gena smn1 dovodo do odsustva sinteze funkcionalnog proteina
Najčešći oblik SMA je tip I koji se javlja u prvim mesecima života, ukoliko postoji genetička predispozicija, bolest se može manifestovati kod starije populacije što je mnogo ređe u odnosu na tip I.
Genetičko testiranje je najpouzdanija metoda koja se primenjuje ukoliko osoba ima simptome SMA ili porodičnu anamnezu SMA. Rana dijagnostika prilikom prvih simptoma je od presudnog značaja za početak lečenja, takođe za otkrivanje nosilaca mutacije ukoliko se planira potomstvo.
U Beo-lab laboratorijama postoji mogućnost analize gena koji se dovode u vezu sa ispoljavanjem SMA kroz Adventia test. Adventia test je skrining test, koji otkriva nosioca mutacije na genu smn1 i smn2. Testiranje osoba koje imaju pozitivnu porodičnu anamnezu za SMA otkriva verovatnoću ispoljavanja poremećaja u budućnosti.
Rano testiranje roditelja otvara mogućnost da se unapred informišu o prisustvu mutiranih smn1 i smn2 gena fetusa, a samim tim i o svim zdravstvenim rizicima i potencijalu za razvoj bolesti kod deteta. Testiranje pre začeća odnosi se na testiranje roditelja i dalje informaciju o reproduktivnim opcijama parova. U okviru Adventia testa vrši se uzorkovanje oba roditelja!
Za više informacija o laboratorijskim testovima dostupnim u Beo-lab laboratorijama, možete nam pisati na mail office@beo-lab.rs ili nas pozvati na broj telefona našeg call centra 0113622888
Povezani tekstovi:
Literatura:
https://smauk.org.uk/support-information/about-sma/the-genetics-of-5q-sma/
https://smauk.org.uk/treatments-research/drugs-being-tested-clinical-trials/
https://smauk.org.uk/living-with-sma/
Radovi:
An Update of the Mutation Spectrum of the Survival Motor Neuron Gene (SMN1) in Autosomal Recessive Spinal Muscular Atrophy (SMA), https://pubmed.ncbi.nlm.nih.gov/10679938/
Molecular and Functional Analysis of Intragenic SMN1 Mutations in Patients with Spinal Muscular Atrophy, https://pubmed.ncbi.nlm.nih.gov/15580564/
The Genetics of Spinal Muscular Atrophy: Progress and Challenges –https://pubmed.ncbi.nlm.nih.gov/25413156/
New and Developing Therapies in Spinal Muscular Atrophy: From Genotype to Phenotype to Treatment and Where Do We Stand? , https://pubmed.ncbi.nlm.nih.gov/32392694/
SMA- Spinal Muscular Atrophy: Past, Present, and Future, https://pubmed.ncbi.nlm.nih.gov/33822657/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9292571/
https://pubmed.ncbi.nlm.nih.gov/33792051/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6396106/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3231874/
https://www.jmdjournal.org/article/S1525-1578(20)30053-2/fulltext
https://smauk.org.uk/support-information/about-sma/the-genetics-of-5q-sma/
https://smauk.org.uk/support-information/about-sma/family-planning/
https://smauk.org.uk/living-with-sma/




